Apert Syndrome is a very rare disorder. According to statistics, there are only 300 cases in the record. Keep in mind that Apert Syndrome was first described in 1894. In a period of more than a century report of only 300 cases makes Apert Syndrome quite rare. Moreover, the statistics reveal only one in 65000 births develop Apert Syndrome. Surprisingly, Asia reports the highest number of Apert Syndrome cases.
A case of 9 months old boy was presented to a clinician. The child had bilateral syndactyly of hands and feet and the head was abnormal in shape. He was the second child of nonconsanguineous parents. Both parents and the other sibling were normal. Mother had a normal delivery with no history of infection, drugs, trauma, disease or medical emergency. Moreover, there was no similar case in their family.
Physical Examination Revealed Enough.
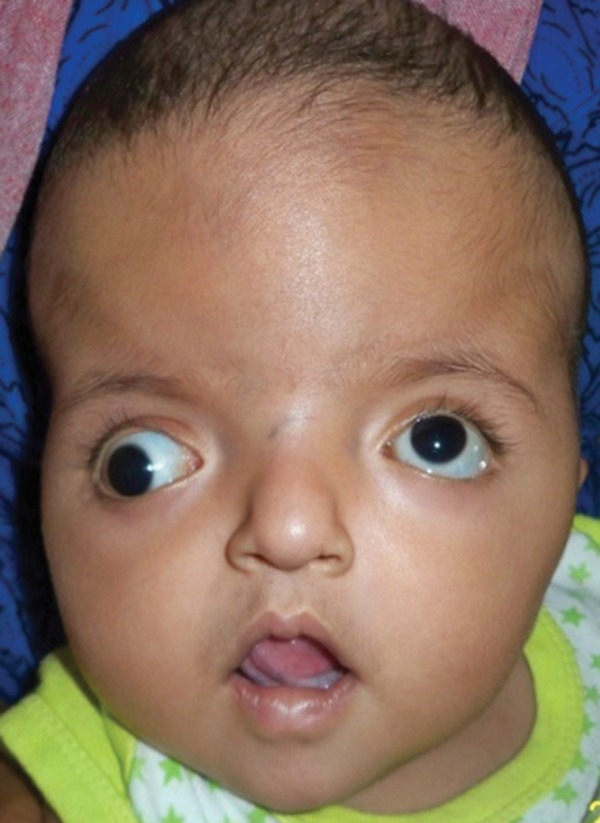
To begin with, the clinician conducted physical examination for close analysis of the child’s skeletal and muscular development. The clinician observed shortened AP which formed abnormal turribrachycephalic head contour. Subsequently, the head had flat occiput and protuberant frontal region. Moreover, the 9 months old child had proptosis, down sliding lateral palpebral fissures, hypertelorism and strabism. The nose was thick. The nasal bridge was depressed and had a bulbous tip at the end. He had to cross bow-shaped lips. The boy had a backward displacement of the maxilla.


After examining the head clinician examined the limbs. The fingers of both hands and feet were completely fused together. However, all fingers had separate nails. In addition, the great toe of both feet was deformed.


Finally, clinician examined the oral cavity. There was a maxillary arch in V shape with a pseudocleft palate.
Radiography Supported Physical Examination.
Radiographs supported the physical examination of the body. According to radiographs, the syndactyly involved only soft tissues. The second, third and fourth phalanges and metacarpals of hands and feet showed synostosis. Radiographs of the skull showed hypertelorism, bitemporal widening, fused coronal sutures, turribrachycephalic skull contour and increased convolutional markings.


In conclusion, the physical examination and radiographs diagnosed the 9 months only boy with Apert Syndrome.
What does Apert Syndrome affect?
Apert Syndrome affects multiple systems at the same time. Here is a list of defects in different organ systems other than those mentioned in the case discussed.
- Out of position anus
- Blocked vagina
- Failed testicles
- Renal dysfunction
- Cardiac dysfunction
- Compromised IQ
- absence of corpus callosum i.e. connects both cerebral hemispheres
- Enlarged brain cavity
- Compromised Autonomic Nervous System (ANS)
Treatment of Apert Syndrome
As far as the treatment of Apert Syndrome is concerned it is a multidisciplinary approach. The treatment begins soon after birth.
- Craniectomy, at the age of 6 months, treats craniosynostosis.
- Corrective surgery, at the age of 1, treats syndactyly. Usually, the surgery takes place various times until the age of 3-4.
- Cosmetic surgery, at the age of 4-6, treats midface deficiency and psuedocleft palate.
- Orthodontic and orthognathic surgery, after the eruption of permanent teeth and their complete growth, treats the deformity in the oral cavity.


